
Two photosynthetic reaction centers are arranged in tandem in photosynthesis of algae and plants
In green algae about eight photons are required (quantum requirement: pho- tons absorbed per molecule O2 produced) for the photosynthetic water split- ting (section 2.4). Instead of the term quantum requirement, one often uses the reciprocal term quantum yield (molecules of O2 produced per photon absorbed). According to the color of irradiated light (action spectrum) the quantum yield dropped very sharply when algae were illuminated with red light above a wavelength of 680 nm (Fig. 3.14). This effect, named “red drop,” remained unexplained since algae contain chlorophyll, which absorbs light at 700 nm. Robert Emerson and coworkers (USA) solved this problem in 1957 when they observed that the quantum yield in the spectral range above 680 nm increased dramatically when algae were illuminated with orange light (650 nm) and red light simultaneously. Then the quantum yield was higher than the sum of both yields when irradiated separately with the light of each wavelength. This Emerson effect led to the conclusion that two differ- ent reaction centers are involved in photosynthesis of green algae (and also of cyanobacteria and higher plants). In 1960 Robert Hill (Cambridge, UK) postulated a reaction scheme (Fig. 3.15) in which two reaction centers are

A
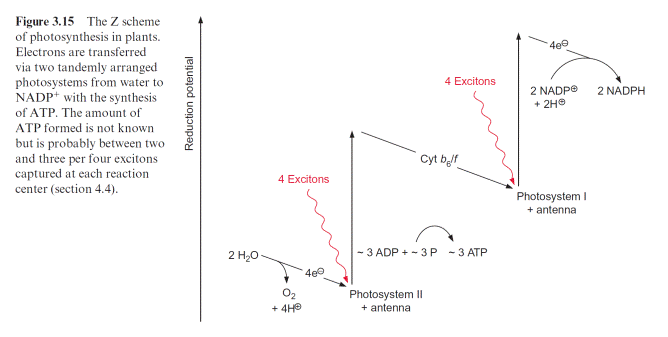


Figure 3.17 Schematic presentation of the localization of the photosynthetic complexes and the H-ATP synthase in the thylakoid membrane. Transport of electrons between PS II and the cytochrome-b6/f complex is mediated by plastohydroquinone (PQH2), and that between the cytochrome-b6/f complex and PS I by plastocyanin (PC). Water splitting occurs on the luminal side of the membrane, and the formation of NADPH and ATP on the stromal side. The electrochemical gradient of protons pumped into the lumen drives ATP synthesis. The number of protons transported to the lumen during electron transport and the proton requirement of ATP synthesis is not known (section 4.4).
Arranged in tandem and connected by an electron transport chain containing cytochrome-b6 and cytochrome-f (cytochrome-f is a cytochrome of the c type; see section 3.7). Light energy of 700 nm was sufficient for the excitation of reaction center I, whereas excitation of the other reaction center II required light of higher energy with a wavelength of 680 nm. The electron flow accord- ing to the redox potentials of the intermediates shows a zigzag, leading to the name Z scheme. The numbering of the two photosystems corresponds to the sequence of their discovery. Photosystem II (PS II) can use light up to a wave- length of 680 nm, whereas photosystem I (PS I) can utilize light with a wave- length up to 700 nm. The sequence of the two photosystems makes it possible that at PS II a very strong oxidant is generated for the oxidation of water and at PS I a very strong reductant is produced for the reduction of NADP (see also Fig. 3.3).
Figure 3.16 gives an overview of electron transport through the photo- synthetic complexes; the carriers of electron transport are drawn according to their electric potential (see also Fig. 3.11). Figure 3.17 shows how the photosynthetic complexes are arranged in the thylakoid membrane. There is a potential difference of about 1.2 volt between the process of water oxi- dation and NADP reduction. The absorbed photons of 680 and 700 nm together correspond to a total potential difference of 3.45 volt (see section 2.2, equation 2.7). Thus, only about one-third of the energy of the pho- tons absorbed by the two photosystems is used to transfer electrons from
water to NADP. In addition to this, about one-eighth of the light energy absorbed by the two photosystems is conserved by pumping protons into the lumen of the thylakoids via PS II and the cytochrome-b6/f complex (Fig. 3.17). This proton transport leads to the formation of a proton gradi- ent between the lumen and the stroma sp ace. An H-ATP synthase, also located in the thylakoid membrane, uses the energy of the proton gradient to synthesize ATP. Thus about half the absorbed light energy of the two photosystems is not used for chemical work but is dissipated as heat. The significance of the loss of energy as heat during photosynthetic electron transport has been discussed in section 2.3.
Water is split by photosystem II
The groups of Horst Witt and Wolfgang Saenger (both in Berlin) resolved the three-dimensional structure of PS II by X-ray structure analysis of crys- tals from the PS II of the thermophilic cyanobacteria Thermosynechococcus elongatis. The subsequent X-ray structure analysis of PS I revealed that PS II and PS I are constructed after the same basic principles as the reaction
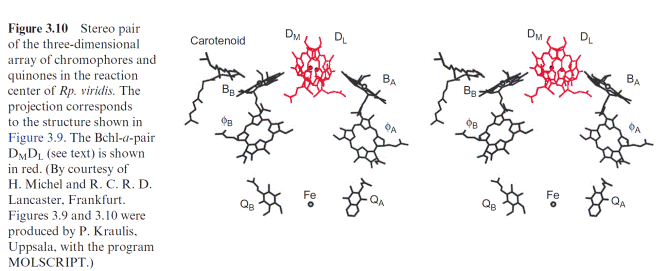
centers of purple bacteria (section 3.4). This, and sequence analyses, clearly demonstrate that all these photosystems have a common origin. Thus PS II also has a chl-a pair in the center, although the distance between the two molecules is so large that probably only one of the two chl-a molecules reacts with the exciton. Two arms, each with one chl-a and one pheophy- tin molecule, are connected with this central pair as in the purple bacteria shown in Figure 3.10. Also in the cyanobacteria, only one of these arms appears to be involved in the electron transport.
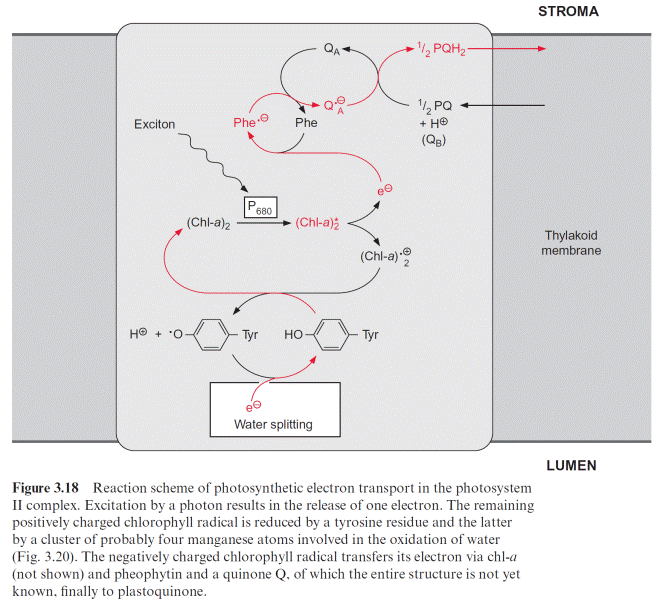
In contrast to the bacterial reaction center the excitation of the reaction center results in an electron transfer via the chl-a monomer to pheophytin (Phe), and from there to a tightly bound plastoquinone (QA), thus forming a semiquinone radical (Fig. 3.18). The electron is then further transferred to a loosely bound plastoquinone (QB). This plastoquinone (PQ) (Fig. 3.19) accepts two electrons and two protons one after the other and is thus reduced to hydroquinone (PQH2). The hydroquinone is released from the photosynthesis complex and may be regarded as the final product of photosystem II. This sequence, consisting of a transfer of a single electron between (chl-a)2 and QA and the transfer of two electrons between QA and QB, corresponds to the reaction sequence shown for Rb. sphaeroides (Fig. 3.11). The only difference is that the quinones are ubiquinone or menaqui- none in bacteria and plastoquinone in photosystem II.
However, the similarity between the reaction sequence in PS II and the photosystem of the purple bacteria applies only to the electron acceptor region. The electron donor function in PS II of plants is completely differ- ent from that in purple bacteria. The electron deficit in (chl-a)2 caused by non-cyclic electron transport is compensated for by electrons derived from the oxidation of water. In the transport of electrons from water to chloro- phyll manganese cations and a tyrosine residue are involved. The (chl-a)2 radical with a redox potential of about 1.1 volt is such a strong oxidant that it can withdraw an electron from a tyrosine residue in the protein of the reaction center and a tyrosine radical remains. This reactive tyrosine residue is often designated as Z. The electron deficit in the tyrosine radical is restored by oxidation of a manganese ion (Fig. 3.20). The PS II com- plex contains several manganese ions, probably four, which are close to each other. This arrangement of Mn ions is called the Mn cluster. The Mn cluster depicts a redox system that can take up and release four electrons. During this process the Mn ions probably change between the oxidation state Mn3 and Mn4. To liberate one molecule of O2 from water, the reaction center must with- draw four electrons and thus capture four excitons. The time differences between the capture of the single exciton in the reaction center depends on the intensity of illumination. If oxidation of water were to proceed stepwise, oxygen radicals could be formed as intermediary products, especially at low light intensities. Oxygen radicals have a destructive effect on biomolecules such as lipids and proteins (section 3.10). The water splitting machinery of the Mn clusters minimizes the formation of oxygen radical intermediates by supplying the reaction center via tyrosine with four electrons one after the other (Fig. 3.20). The Mn cluster is transformed during this transfer from the ground oxidation state stepwise to four different oxidation states (these have been designated as S0 and S1–S4).

Experiments by Pierre Joliot (France) and Bessel Kok (USA) presented evidence that the water splitting apparatus can be in five different oxida- tion states (Fig. 3.21). When chloroplasts that were kept in the dark were then illuminated by a series of light pulses, an oscillation of the oxygen release was observed. Whereas after the first two light pulses almost no O2 was released, the O2 release was maximal after three pulses and then after a further four pulses, and so on. An increasing number of light pulses, how- ever, dampened the oscillation. This can be explained by pulses that do not cause excitation of PS II and thus desynchronize the oscillation. In dark- ened chloroplasts the water splitting apparatus is in the S1 state. After the fourth oxidation state (S4) has been reached, O2 is released in one reaction and the Mn cluster returns to its ground oxidation state (S0). During this reaction, protons from water are released to the lumen of the thylakoids. The formal description of this reaction is:
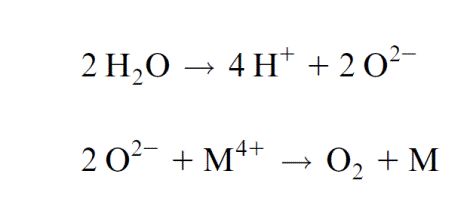
Figuratively speaking, the four electrons needed in the reaction center are loaned in advance by the Mn cluster and then repaid at one stroke by oxidizing water to synthesize one oxygen molecule. In this way the Mn clus- ter minimizes the formation of oxygen radicals in photosystem II. Despite this safety device, still some oxygen radicals are formed in the PS II com- plex which damage the proteins of the complex. The consequences will be discussed in section 3.10.
Photosystem II complex is very similar to the reaction center in purple bacteria
Photosystem II is a complex consisting of at least 20 different subunits (Table 3.2), only two of which are involved in the actual reaction center. For the sake of simplicity the scheme of the PS II complex shown in Fig.
3.22 contains only some of these subunits. The PS II complex is surrounded by an antenna consisting of light harvesting complexes (Fig. 2.13).
The center of the PS II complex is a heterodimer consisting of the sub- units D1 and D2 with six chl-a, two pheophytin, two plastoquinone, and one to two carotenoid molecules bound to it. The D1 and D2 proteins are homologous to each other and also to the L proteins and M proteins from the reaction center of the purple bacteria (section 3.4). As in purple bacteria, only the pheophytin molecule bound to the D1 protein of PS II is involved in electron transport. QA is bound to the D2 protein, whereas QB is bound to the D1 protein. The Mn cluster is probably enclosed by both the D1 and D2 proteins. The tyrosine that is reactive in electron transfer is a constituent
of D1. The subunits O, P, Q stabilize the Mn cluster. The two subunits CP 43 and CP 47 (CP means chlorophyll protein) each bind about 15 chloro- phyll molecules and form the core complex of the antenna shown in Figure
2.10. CP 43 and CP 47 flank both sides of the D1-D2 complex. Cyt-b559 does not seem to be involved in the electron transport of PS II; possibly its func- tion is to protect the PS II complex from light damage. The inner and outer light harvesting complexes of LHC II are arranged at the periphery.
The D1 protein of the PS II complex has a high turnover; it is constantly being resynthesized. It seems that the D1 protein wears out during its func- tion, perhaps through damage by oxygen radicals, which still occurs despite all the protection mechanisms. It has been estimated that the D1 protein is replaced after 106 to 107 catalytic cycles of the PS II reaction center.
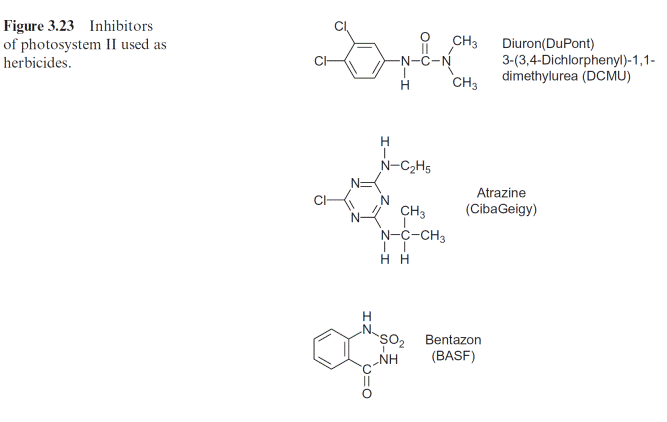
A number of compounds that are similar in their structure to plasto- quinone can block the plastoquinone binding site at the D1 protein, caus- ing inhibition of photosynthesis. Such compounds are used as weed killers (herbicides). Before the effect of these compounds is discussed in detail, some general aspects of the application of herbicides shall be introduced.
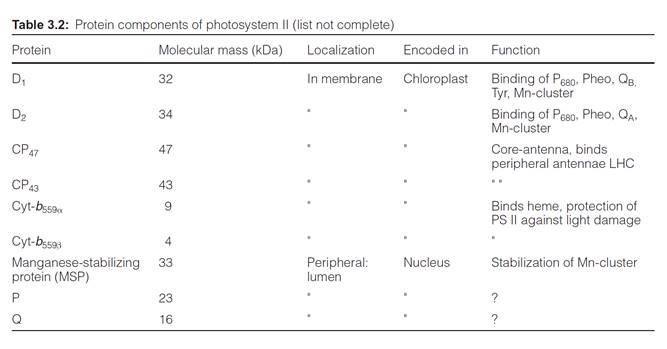
Table 3.4: Protein components of photosystem I (list not complete)

Mechanized agriculture usually necessitates the use of herbicides
About 50% of the money spent worldwide for plant protection is expended for herbicides. The high cost of labor is one of the main reasons for using herbicides in agriculture. It is cheaper and faster to keep a field free of weeds by using herbicides rather than manual labor. Weed control in agriculture is necessary not only to decrease harvest losses by weed competition, but also because weeds hinder the operation of harvesting machineries; fields free of weeds are a prerequisite for a mechanized agriculture. The herbicides usu- ally block a specific reaction of the plant metabolism and have a low toxic- ity for animals and humans. A large number of herbicides (examples will be given at the end of this section) inhibit photosystem II by being antagonists to plastoquinone. To achieve substantial inhibition the herbicide molecule has to bind to most of the many photosynthetic reaction centers. To be effective, 125 to 4,000 g of these herbicides have to be applied per hectare.
In an attempt to reduce the amount of herbicides applied to the soil, new efficient herbicides have been developed that inhibit key biosynthetic processes such as the synthesis of fatty acids, amino acids (sections 10.1 and 10.4), carotenoids, or chlorophyll. There are also herbicides that act as analogues of phytohormones or mitosis inhibitors. Some of these herbi- cides are effective with amounts as low as 5 g per hectare.
Some herbicides are taken up only by the roots and others by the leaves. To keep the railway tracks free of weeds, nonselective herbicides are employed, which destroy the complete vegetation. Nonselective herbicides are also used in agriculture, e.g., to combat weeds in citrus plantations. In the latter case, herbicides are applied that are only taken up by the leaves to combat herbaceous plants at the ground level. Especially interesting are selec- tive herbicides that combat only weeds and effect cultivars as little as possible (sections 12.2 and 15.3). Selectivity can be due to different uptake efficien- cies of the herbicide in different plants, different sensitivities of the metabo- lism towards the herbicide, or different ability of the plants to detoxify the herbicide. Important mechanisms that plants utilize to detoxify herbicides and other foreign compounds (xenobiotics) are the introduction of hydroxyl groups by P-450 monooxygenases (section 18.2) and the formation of glu- tathione conjugates (section 12.2). Selective herbicides have the advantage that weeds can be destroyed at a later growth stage of the cultivars where the dead weeds form a mulch layer conserving water and preventing erosion.
In some cases, the application of herbicides has led to the evolution of herbicide-resistant plant mutants (section 10.4). Conventional breeding has used such mutated plants to generate herbicide-resistant cultivars. In contrast to the occurrence of herbicide resistance by accidental mutations, nowadays genetic engineering is employed on a very large scale to generate cultivars which are resistant to a certain herbicide, allowing weed control in the presence of the growing cultivar (section 22.6)
A large number of herbicides inhibit photosynthesis: the urea deriva- tive DCMU (Diuron, DuPont), the triazine Atrazine (earlier Ciba Geigy), Bentazon (BASF) (Fig. 3.23), and many similar compounds function as herbicides by binding to the plastoquinone binding site on the D1 pro- tein and thus blocking the photosynthetic electron transport. Nowadays, DCMU is not often used, as the required dosage is high and its degradation is slow. It is, however, often used in the laboratory to inhibit photosynthe- sis in an experiment (e.g., of leaves or isolated chloroplasts). Atrazine acts selectively: maize plants are relatively insensitive to this herbicide since they have a particularly efficient mechanism for its detoxification (section 12.2). Because of its relatively slow degradation in the soil, the use of Atrazine has been restricted in some countries, e.g., Germany. In areas where cer- tain herbicides have been used continuously over the years, some weeds have become resistant to these herbicides. In some cases, the resistance can be traced back to mutations resulting in a single amino acid change in the D1-proteins. These changes do not markedly affect photosynthesis of these weeds, but they do decrease binding of the herbicides to the D1-protein.
The cytochrome-b6/f complex mediates electron transport between photosystem II and photosystem I Iron atoms in cytochromes and in iron-sulfur centers have a central function as redox carriers
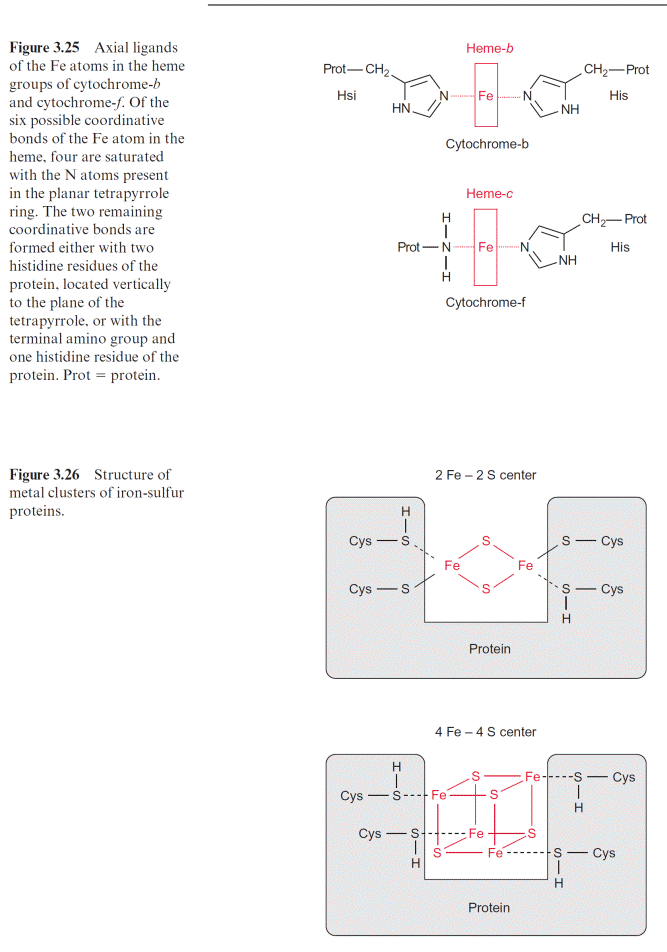
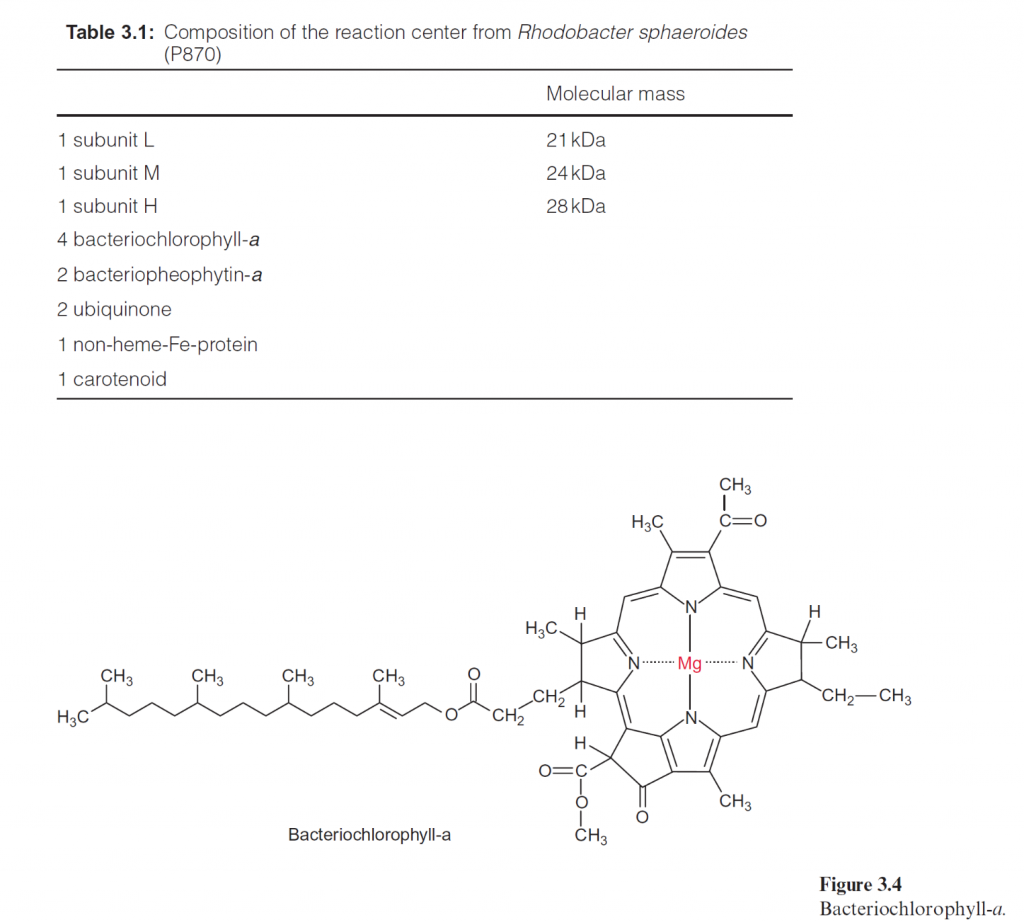
Cytochromes occur in all organisms except a few obligate anaerobes. These are proteins to which one to two tetrapyrrole rings are bound. These tetrapyrroles are very similar to the chromophores of chlorophylls. However, chlorophylls contain Mg as the central atom in the tetrapyr- role, whereas the cytochromes have an iron atom (Fig. 3.24). The tetrapy- rrole ring of the cytochromes with iron as the central atom is called the heme. The bound iron atom can change between the oxidation states Fe and Fe so that cytochromes function as a one-electron-carrier, in con- trast to quinones, NAD(P) and FAD, which transfer two electrons together with protons.
Cytochromes are divided into three main groups, the cytochromes-a, –b, and –c. These correspond to heme-a, –b, and –c. Heme-b may be regarded as the basic structure (Fig. 3.24). In heme-c the -SH-group of a cysteine is added to each of the two vinyl groups of heme-b. In this way heme-c is
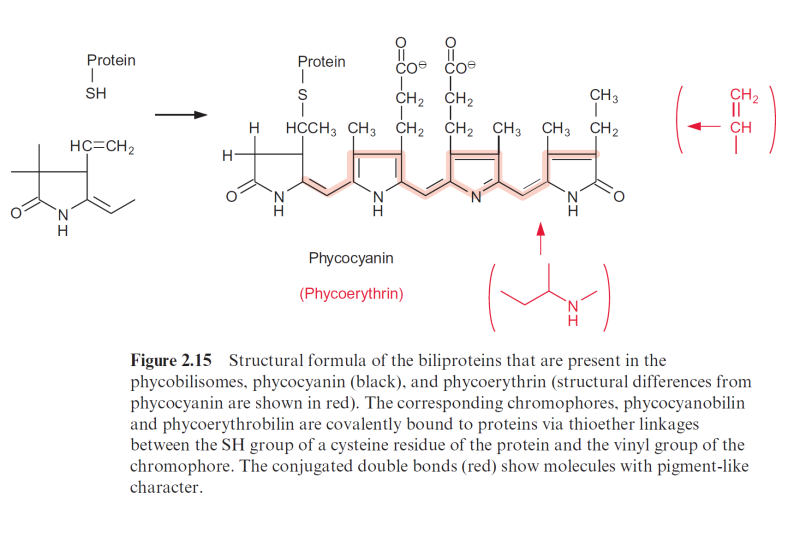
covalently bound by a sulfur bridge to the protein of the cytochrome. Such a mode of covalent binding has already been shown for phycocyanin in Figure 2.15, and there is actually a structural relationship between the correspond- ing apoproteins. In heme-a (not shown) an isoprenoid side chain consisting of three isoprene units is attached to one of the vinyl groups of heme-b. This side chain functions as a hydrophobic membrane anchor, similar to that found in quinones (Figs. 3.5 and 3.19). Heme-a is mentioned here only for the sake of completeness. It plays no role in photosynthesis, but it does have a function in the mitochondrial electron transport chain (section 5.5).

The iron atom in the heme can form up to six coordinative bonds. Four of these bonds are formed with the nitrogen atoms of the tetrapyrrole ring. This ring has a planar structure. The two remaining bonds of the Fe atom coordinate with two histidine residues, which are positioned vertically to the tetrapyrrole plane (Fig. 3.25). Cyt-f (f foliar, in leaves) contains, like cyt-c, one heme-c and therefore belongs to the c-type cytochromes. In cyt-f one bond of the Fe atom coordinates with the terminal amino group of the protein and the other with a histidine residue.
Iron-sulfur centers are of general importance as electron carriers in elec- tron transport chains and thus also in photosynthetic electron transport. Cysteine residues of proteins within iron-sulfur centers (Fig. 3.26) are coor- dinatively or covalently bound to Fe atoms. These iron atoms are linked to each other by S-bridges. Upon acidification of the proteins, the sulfur between the Fe atoms is released as H2S and for this reason it has been called labile sulfur. Iron-sulfur centers occur mainly as 2Fe-2S or 4Fe-4S centers. The Fe atoms in these centers are present in the oxidation states
Fe and Fe. Irrespective of the number of Fe atoms in a center, the oxidized and reduced state of the center differs only by a single charge. For this reason, iron-sulfur centers can take up and transfer only one electron. Various iron-sulfur centers have very different redox potentials, depending on the surrounding protein.

The electron transport by the cytochrome-b6/f complex is coupled to a proton transport
Plastohydroquinone (PQH2) formed by PS II diffuses through the lipid phase of the thylakoid membrane and transfers its electrons to the cytochrome-b6/f complex (Fig. 3.17). This complex then transfers the electrons to plastocy- anin, which is thus reduced. Therefore the cytochrome-b6/f complex has also been called plastohydroquinone-plastocyanin oxidoreductase. Plastocyanin is a protein with a molecular mass of 10.5 kDa, containing a copper atom, which is coordinatively bound to one cysteine, one methionine, and two histidine residues of the protein (Fig. 3.27). This copper atom alternates between the oxidation states Cu and Cu and thus is able to take up and transfer one electron. Plastocyanin is soluble in water and is located in the thylakoid lumen.
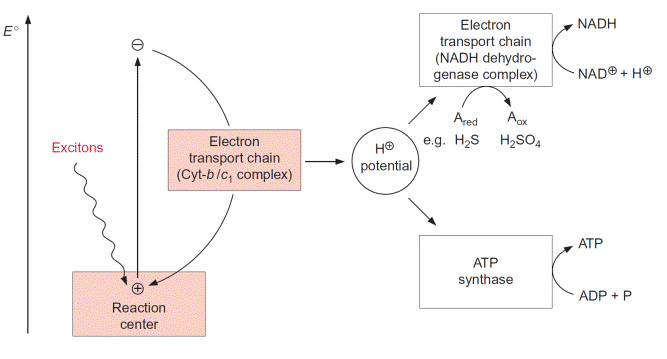
Electron transport through the cyt-b6/f complex proceeds along a poten- tial difference gradient of about 0.4 V (Fig. 3.16). The energy liberated by the transfer of the electron down this redox gradient is conserved by trans- porting protons to the thylakoid lumen. The cyt-b6/f complex is a mem- brane protein consisting of at least eight subunits. The main components of this complex are four subunits: cyt-b6, cyt-f, an iron-sulfur protein called Rieske protein after its discoverer, and a subunit IV. Additionally, there are some smaller peptides and a chlorophyll and a carotenoid of unknown function. The Rieske protein has a 2Fe-2S center with the very positive redox potential of 0.3 V, untypical of such iron-sulfur centers.
The cyt-b6/f complex has an asymmetric structure (Fig. 3.28). Cyt-b6 and subunit IV span the membrane. Cyt-b6 containing two heme-b molecules is almost vertically arranged to the membrane and forms a redox chain across
one iron-sulfur protein. The amino acid sequence of cyt-b in the cyt-b/c1 complex of bacteria and in mitochondria corresponds to the sum of the sequences of cyt-b6 and the subunit IV in the cyt-b6/f complex. Apparently during evolution the cyt-b gene was cleaved into two genes, for cyt-b6 and subunit IV. Whereas in plants the cyt-b6/f complex reduces plastocyanin, the cyt-b/c1 complex of bacteria and mitochondria reduces cyt-c. Cyt-c is a very small cytochrome molecule that is water-soluble and, like plasto- cyanin, transfers redox equivalents from the cyt-b6/f complex to the next complex along the aqueous phase. In cyanobacteria, which also possess a cyt-b6/f complex, the electrons are transferred from this complex to photo- system I via cyt-c instead of plastocyanin. The great similarity between the cyt-b6/f complex in plants and the cyt-b/c1 complexes in bacteria and mito- chondria suggests that these complexes have basically similar functions in photosynthesis and in mitochondrial oxidation: they are proton transloca- tors that are driven by a hydroquinone-plastocyanin (or -cyt-c) reductase.
The interplay of PS II and the cyt-b6/f complex electron transport causes the transport of protons from the stroma space to the thylakoid lumen. The principle of this transport is explained in the schematic presentations of Figures 3.28 and 3.29. A crucial point is that the reduction and oxida- tion of the quinone occur at different sides of the thylakoid membrane. The required protons for the reduction of PQ (Qb) by the PS II complex are taken up from the stroma space. Subsequently PQH2 diffuses across the lipid phase of the membrane to the binding site in the lumenal region of the cyt-b6/f complex where it is oxidized by the Rieske protein and cyt-f to yield reduced plastocyanin. The protons of this reaction are released into the thylakoid lumen. According to this scheme, the capture of four excitons by the PS II complex transfers four protons from the stroma space to the lumen. In addition four protons produced during water splitting by PS II are released into the lumen as well.


The number of protons pumped through the cyt-b6/f complex can be doubled by a Q-cycle
Studies with mitochondria indicated that during electron transport through the cyt-b/c1 complex, the number of protons transferred per transported electron is larger than four (Fig. 3.29). Peter Mitchell (Great Britain), who established the chemiosmotic hypothesis of energy conservation (section 4.1), also postulated a so-called Q-cycle, by which the number of trans- ported protons for each electron transferred through the cyt-b/c1 complex is doubled. It later became apparent that the Q-cycle also has a role in pho- tosynthetic electron transport.
Figure 3.30 shows the principle of Q-cycle operation in the photosyn- thesis of chloroplasts. The cyt–b6/f complex contains two different bind- ing sites for conversion of quinones, one located at the stromal side and the other at the luminal side of the thylakoid membrane (Fig. 3.28). The plastohydroquinone (PQH2) formed in the PS II complex is oxidized by the Rieske iron-sulfur center at the binding site adjacent to the lumen. Due to its very positive redox potential, the Rieske protein tears off one electron from the plastohydroquinone. Because its redox potential is very negative, the remaining semiquinone is unstable and transfers its electron to the first heme-b of the cyt-b6 (bp) and from there to the other heme-b (bn), thus rais- ing the redox potential of heme bn to about –0.1 V. In this way a total of four protons are transported to the thylakoid lumen per two molecules of plastohydroquinone oxidized. Of the two plastoquinone molecules (PQ) formed, only one molecule returns to the PS II complex.
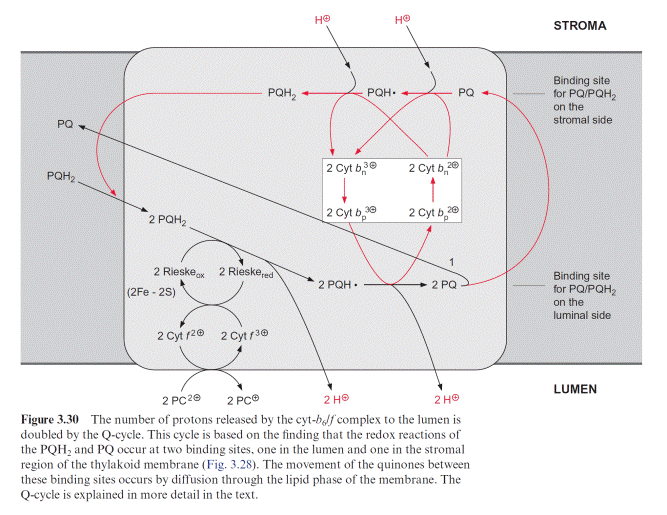
The other PQ dif- fuses away from the cyt-b6/f complex through the lipid phase of the mem- brane to the stromal binding site of the cyt-b6/f complex to be reduced via semiquinone to hydroquinone by the high reduction potential of heme-bn. This is accompanied by the uptake of two protons from the stromal space. The hydroquinone thus regenerated diffuses through the membrane back to the luminal binding site where it is oxidized in turn by the Rieske protein, and so on. In total, the number of transported protons is doubled by the Q-cycle (1/2 1/4 1/8 1/16 1/n 1). The fully operating Q-cycle transports four electrons through the cyt-b6/f complex which results in total to the transfer of eight protons from the stroma to the lumen. The func- tion of this Q-cycle in mitochondrial oxidation is now undisputed, while its function in photosynthetic electron transport is still a matter of contro- versy. The analogy of the cyt-b6/f complex to the cyt-b/c1 complex suggests that the Q-cycle also plays an important role in chloroplasts. So far, the operation of a Q-cycle in plants has been observed mainly under low light conditions. The Q-cycle is perhaps suppressed by a high proton gradient generated across the thylakoid membrane, for instance, by irradiation with high light intensity. In this way the flow of electrons through the Q-cycle could be adjusted to the energy demand of the plant cell.
Photosystem I reduces NADP
Plastocyanin that has been reduced by the cyt-b6/f complex diffuses through the lumen of the thylakoids, binds to a positively charged binding site of PS I, transfers its electron, and the resulting oxidized form diffuses back to the cyt-b6/f complex (Fig. 3.31).
Also the reaction center of PS I with an absorption maximum of 700 nm contains a chlorophyll pair (chl-a)2 (Fig. 3.31). As in PS II, the excitation caused by a photon reacts probably with only one of the two chlorophyll molecules. The resulting (chl-a)2 is then reduced by plastocyanin. It is assumed that (chl-a)2 transfers its electron to a chl-a monomer (A0), which then transfers the electron to a strongly bound phylloquinone (Q) (Fig. 3.32). Phylloquinone contains the same phytol side chain as chl-a and its function corresponds to QA in PS II. The electron is transferred from the semiphylloquinone to an iron-sulfur center named FX. FX is a 4Fe-4S center

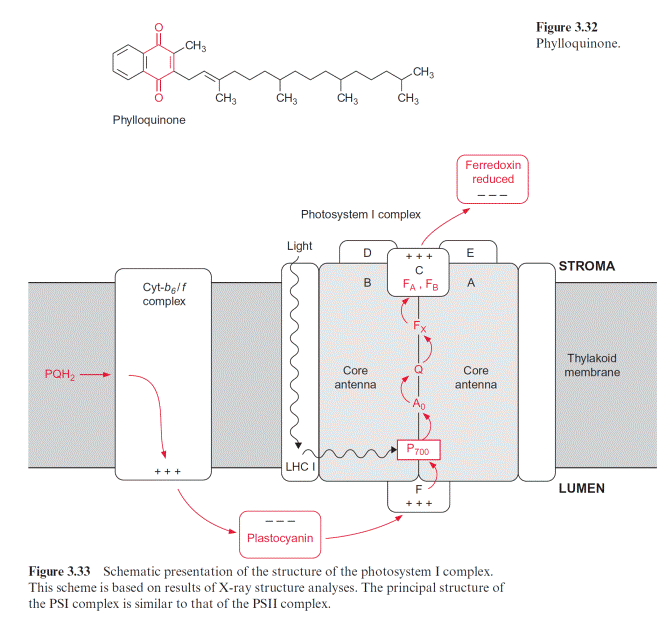
with a very negative redox potential. It transfers one electron to two other 4Fe-4S centers (FA, FB), which in turn reduce ferredoxin, a protein with a molecular mass of 11 kDa with a 2Fe-2S center. Ferredoxin also takes up and transfers only one electron. The reduction occurs at the stromal side of the thylakoid membrane. For this purpose, the ferredoxin binds at a posi- tively charged binding site on subunit D of PS I (Fig. 3.33). The reduction of NADP by ferredoxin, catalyzed by ferredoxin-NADP reductase, yields NADPH as an end product of the photosynthetic electron transport.
The PS I complex consists of at least 17 different subunits, of which some are shown in Table 3.4. The center of the PS I complex is a heterodimer (as is the center of PS II) consisting of subunits A and B (Fig. 3.33). The molec- ular masses of A and B (each 82–83 kDa) correspond approximately to the sum of the molecular masses of the PS II subunits D1 and CP43, and D2 and CP47, respectively (Table 3.2). In fact, both subunits A and B have a dou- ble function. Like D1 and D2 in PS II, they bind chromophores (chl-a) and redox carriers (phylloquinone, FeX) of the reaction center and, additionally, they contain about 100 chl-a molecules as antennae pigments. Thus, the het- erodimer of A and B represents the reaction center and the core antenna as well. The three-dimensional structure of photosystem I in cyanobacteria, green algae and plants has been resolved. The principal structure of pho- tosystem I, with a central pair of chl-a molecules and two branches, each with two chlorophyll molecules, is very similar to photosystem II and to the bacterial photosystem (Fig. 3.10). It has not been definitely clarified whether both or just one of these branches are involved in the electron transport. The Fe-S-centers FA and FB are ascribed to subunit C, and subunit F is con- sidered to be the binding site for plastocyanin.


The light energy driving the cyclic electron transport of PS I is only utilized for the synthesis of ATP
Besides the noncyclic electron transport discussed so far, cyclic electron transfer can also take place in which the electrons from the excited pho- tosystem I are transferred back to the ground state of PS I, probably via the cyt-b6/f complex (Fig. 3.34). The energy thus released is used only for the synthesis of ATP, and NADPH is not formed. This electron transport is termed cyclic photophosphorylation. In intact leaves, and even in iso- lated intact chloroplasts, it is quite difficult to differentiate experimentally between cyclic and non-cyclic photophosphorylation. It has been a matter of debate as to whether and to what extent cyclic photophosphorylation occurs in a leaf under normal physiological conditions. Recent evaluations of the proton stoichiometry of photophosphorylation (see section 4.4) sug- gest that the yield of ATP in noncyclic electron transport is not sufficient for the requirements of CO2 assimilation, and therefore cyclic photophos- phorylation seems to be required to synthesize the lacking ATP. Moreover, cyclic photophosphorylation must operate at very high rates in the bundle sheath chloroplasts of certain C4 plants (section 8.4). These cells have a high demand for ATP and they contain high PS I activity but very little PSPresumably, the cyclic electron flow is governed by the redox state of theacceptor of the photosystem in such a way that by increasing the reduction of the NADP system, and consequently of ferredoxin, the diversion of the
electrons in the cycle is enhanced. The function of cyclic electron transport is probably to adjust the rates of ATP and NADPH formation according to the plant’s demand.
Despite intensive investigations, the pathway of electron flow from PS I to the cyt-b6/f complex in cyclic electron transport remains unresolved. It has been proposed that cyclic electron transport is structurally separated from the linear electron transport chain in a super complex. Most experi- ments on cyclic electron transport have been carried out with isolated thy- lakoid membranes that catalyze only cyclic electron transport when redox mediators, such as ferredoxin or flavin adenine mononucleotide (FMN, Fig. 5.16), have been added. Cyclic electron transport is inhibited by the antibiotic antimycin A. It is not clear at which site this inhibitor functions. Antimycin A does not inhibit noncyclic electron transport.
Surprisingly, proteins of the NADP dehydrogenase complex of the mitochondrial respiratory chain (section 5.5) have also been identified in the thylakoid membrane of chloroplasts. The function of these proteins in chloroplasts is still not known. The proteins of this complex occur very fre- quently in chloroplasts from bundle sheath cells of C4 plants, which have little PS II but a particularly high cyclic photophosphorylation activity (section 8.4). These observations raise the possibility that in cyclic electron transport the flow of electrons from NADPH or ferredoxin to plastoqui- none proceeds via a complex similar to the mitochondrial NADH dehydro- genase complex. As will be shown in section 5.5, the mitochondrial NADH dehydrogenase complex transfers electrons from NADH to ubiquinone. Results indicate that an additional pathway for a cyclic electron transport exists in which electrons are directly transferred via a plastoquinone reduct- ase from ferredoxin to plastoquinone.
In the absence of other acceptors electrons can be transferred from photosystem I to oxygen
When ferredoxin is very highly reduced, it is possible that electrons are
transferred from PS I to oxygen to form superoxide radicals (•O) (Fig.
3.35). This process is called the Mehler reaction. The superoxide radical reduces metal ions present in the cell such as Fe3 and Cu2 (Mn):
The hydroxyl radical (•OH) is a very aggressive substance and damages enzymes and lipids by oxidation. The plant cell has no protective enzymes against •OH. Therefore it is essential that a reduction of the metal ions be prevented by rapid elimination of •O by superoxide dismutase. But hydrogen peroxide (H2O2) also has a damaging effect on many enzymes. To prevent such damage, hydrogen peroxide is eliminated by an ascorbate
peroxidase located in the thylakoid membrane. Ascorbate, an important anti- oxidant in plant cells (Fig. 3.36), is oxidized by this enzyme and converted to the radical monodehydroascorbate, which is spontaneously reconverted by photosystem I to ascorbate via reduced ferredoxin. Monodehydroascorbate can be also reduced to ascorbate by an NAD(P)H-dependent monodehy- droascorbate reductase that is present in the chloroplast stroma and the cytosol.
As an alternative to the preceding reaction, two molecules of mono- dehydroascorbate can dismutate to ascorbate and dehydroascorbate. Dehydroascorbate is reconverted to ascorbate by reduction with glutath- ione in a reaction catalyzed by dehydroascorbate reductase present in the stroma (Fig. 3.37). Glutathione (GSH) occurs as an antioxidant in all plant
cells (section 12.2). It is a tripeptide composed of the amino acids glutamate, cysteine, and glycine (Fig. 3.38). Oxidation of GSH results in the forma- tion of a disulfide (GSSG) between the cysteine residues of two glutathione molecules. Reduction of GSSG is catalyzed by a glutathione reductase with NADPH as the reductant (Fig. 3.37).
The major function of the Mehler-ascorbate-peroxidase cycle is to dis- sipate excessive excitation energy of photosystem I as heat. The absorption of a total of eight excitons via PS I results in the formation of two super- oxide radicals and two molecules of reduced ferredoxin, the latter serving as a reductant for eliminating H2O2 (Fig. 3.35). The transfer of electrons to oxygen by the Mehler reaction is a reversal of the water splitting of PS
II. As will be discussed in the following section, the Mehler reaction occurs when ferredoxin is very highly reduced. The only gain of this reaction is the generation of a proton gradient from electron transport through PS II and the cyt-b6/f complex. This proton gradient can be used for the synthesis of ATP if ADP is present. But since there is usually a shortage in ADP under the conditions of the Mehler reaction, it mostly results in the formation of a high pH gradient. A feature common to the Mehler reaction and cyclic electron transport is that there is no net production of NADPH. For this reason, electron transport via the Mehler reaction has been termed pseudo- cyclic electron transport.
Yet another group of antioxidants was recently found in plants, the so- called peroxiredoxins. These proteins, comprising -SH groups as redox car- riers, have been known in the animal world for some time. Ten different peroxiredoxin genes have been identified in the model plant Arabidopsis. Peroxiredoxins, being present in chloroplasts as well as in other cell com- partments, differ from the aforementioned antioxidants glutathione and ascorbate in that they reduce a remarkably wide spectrum of peroxides, such as H2O2, alkylperoxides, and peroxinitrites. In chloroplasts, oxidized peroxiredoxins are reduced by photosynthetic electron transport of photo- system I with ferredoxin and thioredoxin as intermediates.
Instead of ferredoxin, PS I can also reduce methylviologen. Methylviologen, also called paraquat, is used commercially as a herbicide
(Fig. 3.39). The herbicidal effect is due to the reduction of oxygen to super- oxide radicals. Additionally, paraquat competes with dehydroascorbate for the reducing equivalents provided by photosystem I. Therefore, in the pres- ence of paraquat, ascorbate is no longer regenerated from dehydroascor- bate and the ascorbate peroxidase reaction can no longer proceed. The increased production of superoxide and decreased detoxification of hydro- gen peroxide in the presence of paraquat causes severe oxidative damage to mesophyll cells, noticeable by a bleaching of the leaves. In the past, paraquat has been used to destroy marijuana fields in South America.
Regulatory processes control the distribution of the captured photons between the two photosystems
Linear photosynthetic electron transport through the two photosystems requires the even distribution of the captured excitons between them. As discussed in section 2.4, the excitons are transferred preferentially to the chromophore which requires the least energy for excitation. Photosystem I (P700) being on a lower energetic level than PS II (Fig. 3.16) requires less energy for excitation than photosystem II (P680). In an unrestricted compe- tition between the two photosystems, excitons would primarily be directed to PS I. Due to this imbalance, the distribution of the excitons between the two photosystems must be regulated. The spatial separation of PS I and PS II and their antennae in the thylakoid membrane plays an important role in this regulation.
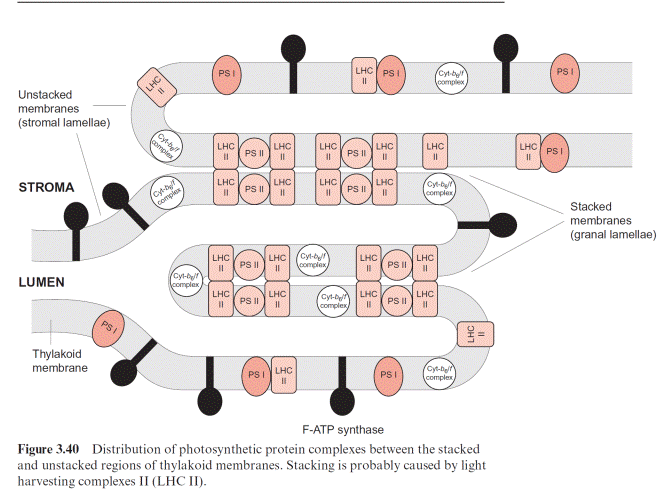
In chloroplasts, the thylakoid membranes are present in two differ- ent arrays, as stacked and unstacked membranes. The outer surface of the unstacked membranes has free access to the stromal space; these mem- branes are called stromal lamellae (Fig. 3.40). In the stacked membranes, the neighboring thylakoid membranes are in direct contact with each other. These membrane stacks can be seen as grains (grana) in light microscopy and are therefore called granal lamellae.
ATP synthase and the PS I complex (including its light harvesting com- plexes, not further discussed here) are located either in the stromal lamellae or in the outer membrane region of the granal lamellae. Therefore, these proteins have free access to ADP and NADP in the stroma. The PS II complex, on the other hand, is primarily located in the granal lamellae. Peripheral LHC II subunits attached to the PS II complex (section 2.4) contain a protein chain protruding from the membrane, which can prob- ably interact with the LHC II subunit of the adjacent membrane and thus
cause tight membrane stacking. The cyt-b6/f complexes are only present in stacked membranes. Since the proteins of PS I and F-ATP-synthase project into the stroma space, they do not fit into the space between the stacked membranes. Thus the PS II complexes in the stacked membranes are sepa- rated spatially from the PS I complexes in the unstacked membranes. It is assumed that this prevents an uncontrolled spillover of excitons from PS II to PS I.
However, the spatial separation of the two photosystems and thus the spillover of excitons from PS II to PS I can be regulated. For example, if the excitation of PS II is greater than that of PS I, plastohydroquinone accumulates, which cannot be oxidized rapidly enough via the cyt-b6/f com- plex by PS I. Under these conditions, a protein kinase is activated, which phosphorylates the hydroxyl groups of threonine residues of peripheral LHC II subunits, causing a conformational change of the LHC protein. As a result of this, the affinity to PS II is decreased and the LHC II subunits dissociate from the PS II complexes. Furthermore, due to the changed con- formation, LHC II subunits can now bind to PS I, mediated by the H sub- unit of PS II. This LHC II-PS I complex purposely increases the spillover of excitons from LHC II to PS I. In this way the accumulation of reduced plastoquinone decreases the excitation of PS II and enhances the excitation of PS I. A protein phosphatase facilitates the reversal of this regulation. This regulatory process, which has been simplified here, enables an opti- mized distribution of the captured photons between the two photosystems, independent of the spectral quality of the absorbed light.
Excess light energy is eliminated as heat
Plants face the general problem that the energy of irradiated light can be much higher than the demand of photosynthetic metabolites such as NADPH and ATP. This is the case when very high light intensities are present and the metabolism cannot keep pace. Such a situation arises at low temperatures, when the metabolism is slowed down because of decreased enzyme activities (cold stress) or at high temperatures, when stomata close to prevent loss of water. Excess excitation of the photosystems could result in an excessive reduction of the components of the photosynthetic electron transport.
Very high excitation of photosystem II, recognized by the accumulation of plastohydroquinone, results in damage to the photosynthetic appara- tus, termed photoinhibition. A major cause of this damage is an overexcita- tion of the reaction center, by which chlorophyll molecules attain a triplet state, resulting in the formation of aggressive singlet oxygen (section 2.3). The damaging effect of triplet chlorophyll can be demonstrated by placing
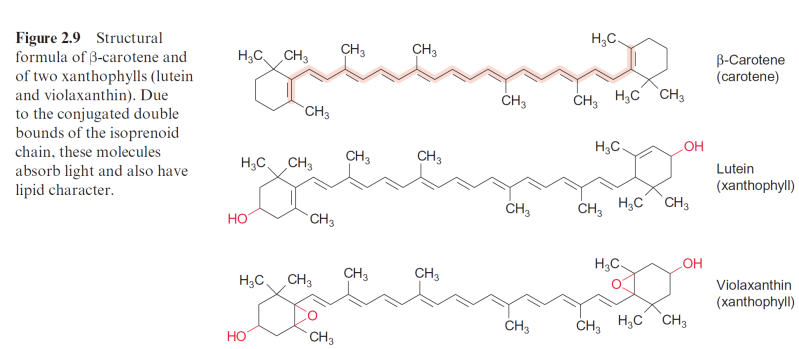
a small amount of chlorophyll under the human skin, which after illumina- tion causes severe tissue damage. This photodynamic principle is utilized in medicine for the selective therapy of skin cancer. Carotenoids (e.g., –carotene, Fig. 2.9) are able to convert the triplet state of chlorophyll and the singlet state of oxygen to the corresponding ground states by forming a triplet carotenoid, which dissipates its energy as heat. In this way carotenoids have an important protective function. If under certain conditions this protective function of carotenoids is una- ble to cope with excessive excitation of PS II, the remaining singlet oxy- gen has a damaging effect on the PS II complex. The site of this damage could be the D1 protein of the photosynthetic reaction center in PS II, which already under normal photosynthetic conditions experiences a high turnover (see section 3.6). When the rate of D1-protein damage exceeds the rate of its resynthesis, the rate of photosynthesis is decreased, resulting in photoinhibition.
Plants have developed several mechanisms to protect the photosynthetic apparatus from light damage. One mechanism is chloroplast avoidance movement, in which chloroplasts move under high light conditions from the cell surface to the side walls of the cells. Another way is to dissipate the energy arising from an excess of excitons as heat. This process is termed nonphotochemical quenching of exciton energy. Although our knowledge of this quenching process is still incomplete, it is undisputed that zeaxan- thin plays an important role. Zeaxanthin causes the dissipation of exciton energy to heat by interacting with a chlorophyll-binding protein (CP 22) of photosystem II. Zeaxanthin is formed by the reduction of the diepoxide vio- laxanthin. The reduction proceeds with ascorbate as the reductant and the monoepoxide antheraxanthin is formed as an intermediate. Zeaxanthin can be reconverted to violaxanthin by epoxidation which requires NADPH and O2 (Fig. 3.41). Formation of zeaxanthin by diepoxidase takes place on the luminal side of the thylakoid membrane at an optimum pH of 5.0, whereas the regeneration of violaxanthin by the epoxidase proceeding at the stro- mal side of the thylakoid membrane occurs at about pH 7.6. Therefore, the formation of zeaxanthin requires a high pH gradient across the thylakoid membrane. As discussed in connection with the Mehler reaction (section 3.9), a high pH gradient can be an indicator of the high excitation state of photosystem II. When there is too much excitation energy, an increased pH gradient initiates zeaxanthin synthesis, dissipating excess energy of the PS II complex as heat. This mechanism explains how under strong sunlight most plants convert 50% to 70% of all the absorbed photons to heat. The non-photochemical quenching of excitation energy is the primary way for plants to protect themselves from too much light energy. In comparison, the Mehler reaction (section 3.9) and photorespiration (section 7.7) under

Figure 3.35 A schemefor the Mehler reaction.Upon strong reduction offerredoxin, electrons aretransferred by the Mehlerreaction to oxygen andsuperoxide is formed. Theelimination of this highlyaggressive superoxideradical involves reactionscatalyzed by superoxidedismutase and ascorbateperoxidase


elker and Or: Soil Hydrology and Biophysics



































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}