
Ion Exchange in Soil: Cation and Anion
Cation Exchange:
In a near neutral soil, calcium remains adsorbed on colloidal particle. Hydrogen ion (H+ ) generated as organic and mineral acids formed due to decomposition of organic matter. In colloid, hydrogen ion is adsorbed more strongly than is the calcium (Ca++). The reaction takes place rapidly and the interchange of calcium and hydrogen is Chemically equivalent.
The reaction is as follows:

{kind=link}
This phenomenon of the exchange of cations between soil and salt solution is known as Cation exchange or Base exchange and the cations that take part in this reaction are called exchangeable cations. Cation exchange reactions are reversible.
Hence, if some form of limestone or other basic calcium compound is applied to an acid soil, the reverse of the replacement just given above occurs. The active calcium ions replace the hydrogen and other cations by mass action. As a result, the clay becomes higher in exchangeable calcium and lower in adsorbed hydrogen and aluminium.
======================================
Cation preferences
When the valence of the cations are equal (i.e. both +1 charge) the cation with the smallest hydrated radius is more strongly adsorbed. In the case of the monovalent cations of potassium and sodium, the potassium ion is more strongly adsorbed since it has a smaller hydrated radius and hence is more strongly adsorbed to the site of the negative charge. In comparison the sodium ion is so loosely held and so ready to hydrate that sodium rich soil will disperse.
| Radius | Unit | Na+ | K+ | Mg2+ | Ca2+ | Al3+ |
| Non-hydrated | nm | 0.095 | 0.133 | 0.066 | 0.099 | 0.050 |
| Hydrated | nm | 0.360 | 0.330 | 0.430 | 0.410 | 0.480 |
This is similarly the case with the divalent cations of calcium and magnesium. Because the hydrated magnesium ion is larger than that of calcium, the magnesium ion is held more weakly and behaves in some instances in soil (i.e. when calcium is low) like sodium.
The charge of the cation and the size of the hydrated cation essentially govern the preferences of cation exchange equilibria. In summary, highly charged cations tend to be held more tightly than cations with less charge and secondly, cations with a small hydrated radius are bound more tightly and are less likely to be removed from the exchange complex. The combined influence of these two criteria can be summarized generally by the lysotrpoic series.

aluminium > calcium > magnesium > potassium, ammonium-NH4+ > sodium > hydrogen
It indicates, from left to right, the decreasing strength of adsorption of the various cations. As such, the less tightly held cations are located furthest from the surface of colloids and are most likely to be leached away or further down the profile most quickly. Conversely, the most strongly adsorbed cations will tend to move the slowest down through the profile.
The proportion and kinds of cations adsorbed on soil mineral particles and organic colloids is also a function of the concentration of cations in the soil solution. If the concentration of a cation in soil solution is high, there is an increased chance or tendency for that cation to be adsorbed.
This is the reason that dissolved gypsum (CaSO4) is added to ameliorate sodic soil. In this case, the addition of dissolved gypsum increases the concentration of calcium in the soil solution and this leads to an increase in calcium ions on the exchange complex at the expense of exchangeable sodium.
The major source of cations in soil solution are from mineral weathering (i.e. primary minerals), mineralization of organic matter and addition of soil ameliorants (i.e. lime, gypsum, etc).
Soil Properties: Exchangeable Cations
======================================
If a soil is treated with a liberal application of a fertilizer containing potassium chloride, following reaction may occur:

{kind=link}
Some of the added potassium pushes its way into the colloidal complex and forces out equivalent quantities of calcium, hydrogen and other elements (e.g., M) which appear in the soil solution. The adsorption of the added potassium largely in an available condition. Hence, cation exchange is an important consideration for making already present nutrients in soils available to plants. Cation exchange also makes available the nutrients, applied in commercial fertilizers form.
Cation Exchange Capacity (C.E.C.):
The cation exchange capacity of a soil represents the capacity of the colloidal complex to exchange all its cations with the cations of the electrolyte solution (surrounding liquid). It also represents the total cation adsorbing capacity of a soil. Cation exchange in most soils increases with pH. At a very low pH value, C.E.C. is higher and at high pH, C.E.C. is relatively lower.
Factors affecting the Cation Exchange Capacity:
The following factors affect the cation exchange capacity:
(i) Soil texture:
Fine-textured (clay) soils tend to have higher cation exchange capacity (CEC) than sandy soils. Cation exchange capacity for clay soils usually exceeds 30 me/100 gm. while the value ranges from 0 to 5 for sandy soils.
(ii) Organic matter content:
Organic matter content of a soil affects the CEC. Higher organic matter content in a soil have higher CEC.
(iii) Amount and kind of clay:
Montmorillonite has higher CEC in comparison to illite or kaolinite clay.
(iv) pH:
The cation exchange capacity of most soils increases with pH. At very low pH value, the cation exchange capacity is also generally low. As the pH is raised, the negative charges on some 1 : 1 type silicate clay (Kaolinite), humus and Fe, Al oxides increases, thereby increasing the cation exchange capacity.
Milliequivalents:
The cation exchange capacity (C.E.C.) is expressed in terms of equivalents or more specifically, as milliequivalents per 100 grams. The term equivalent is defined as one gram atomic weight of hydrogen (or the amount of any other ion) that will combine with or displace this amount of hydrogen. For monovalent ions such as Na+, K+, NH4+ and CI–, the equivalent weight and atomic weight are the same since they can replace one H+ ion. Divalent cations such as Ca++ and Mg++ can take the place of two H+ ions.
The milliequivalent weight of a substance is one thousandth of its atomic weight. Since the equivalent weight of hydrogen is about 1 gm., the term milliequivalent (meq) may be defined as 1 milligram of hydrogen. It indicates that other ions also may be expressed in terms of milliequivalents.
Consider calcium, for example. Ca has an atomic weight of 40 compared to 1 for hydrogen. Each Ca++ ion has two charges and is thus, equivalent to two H+ ions. Therefore, the amount of calcium required to displace 1 mg of hydrogen is 40/2 = 20 mg (atomic wt. divided by 2 to obtain the equivalent wt.). This is the weight of 1 meq of calcium.
If 100 grams of a certain clay is capable of exchanging a total of 250 meq of calcium, the cation exchange capacity is 250/20 = 12.5 meq per 100 gm. The milliequivalent method of expression can be converted easily to practical field terms. For example, 1 meq of hydrogen can be replaced on the colloids by 1 meq of CaCO3(limestone). The molecular weight of CaCO3 is 100, it contains 2 equivalent weights (divalent).
Since the amount of CaCC3 needed is only 1 meq wt., 100/2 = 50 mg will be needed to replace 1 mg of hydrogen (or 1 meq). In view of fact that 1 meq of H+ per 100 grams can be expressed as 20 pounds of hydrogen per grams of soil.
Expressed in the metric system this figure is 1100 kilograms per hectare. In general, the more clay there is in a soil, the higher the C.E.C. Sandy soils have, on the average 0.5 m.e. of C.E.C. per 100 gm. of soil, while for clay soils, it usually exceeds 30 m.e./100 gm.
Percentage Base Saturation of Soils:
Hydrogen and aluminium tend to dominate acid soils, both contributing to the concentration of H+ ions in the soil solution. Adsorbed hydrogen contributes directly to the H+ ion concentration in the soil. A+++ions do so indirectly through hydrolysis.
======================================
Base Saturation – Calculating Cation Exchange Capacity, Base Saturation, and Calcium Saturation
Base saturation is calculated as the percentage of CEC occupied by base cations. Figure 2 shows two soils with the same CEC, but the soil on the right has more base cations (in blue). Therefore, it has a higher base saturation. Base saturation is closely related to pH; as base saturation increases, pH increases.
Base Saturation (%) = (Base cations/CEC) x 100
Similarly, we can calculate the base saturation for each individual base cation. Calcium base saturation is calculated as the percentage of CEC occupied by calcium cations. In Figure 2, the soil on the right has twice as many calcium cations (Ca2+), thus a higher calcium saturation.
Calcium Saturation (%) = (Calcium cations/CEC) x 100

======================================
Reactions are as follows:
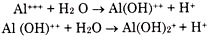
{kind=link}
Most of the other cations (Ca++, Mg++), called exchangeable bases, neutralize soil acidity. The proportion of the cation exchange capacity (C.E.C.) occupied by these bases is called the percentage base saturation. Thus, if the % base saturation is 80 in clay loam soil, 4/5th of the cation exchange capacity (20 meq) is satisfied by bases, the other by hydrogen and aluminium. Same as, 50% base saturation in clay soil having 20 meq C.E.C. x 1/2 (10 meq C.E.C.).of the C.E.C. is satisfied by bases likewise in sandy loam soil with a C.E.C. of only 10 meq, 80% base saturation satisfied, 4/5 of C.E.C.
A definite correlation exists between the percentage base saturation of a soil and its pH. As the base saturation is reduced as a result of loss of calcium in drainage, the pH is also lowered (more acidity)in a definite proportion.
Within the range pH 5 to 6, the ratio for humid temperate region mineral soils is roughly at 5% base saturation, change for every 0.10 change in pH. thus, if the percentage base saturation is 50% at pH 5.5, it should be 25% and 75% at pH 5.0 and 6.0.
Role of Cation Exchange:
Importance of exchangeable cations on plant nutrients is discussed below:
Cation exchange reactions are very important chemical reactions for the availability of plant nutrients in the soil. The capacity of soil to exchange cations is the best single index of soil fertility. Plant roots, when they come in contact with colloidal particles, absorb exchangeable cations directly by inter-exchange or contact exchange between the root hairs and colloidal complex.
(a) Nature and content of exchangeable bases:
The nature and content of exchangeable bases in a soil have an important bearing on its general properties. In all normal fertile soils the total exchangeable bases (Ca, Mg, K, Na) constitute about 80 to 90% of the cation adsorbing capacity. Exchangeable hydrogen is usually under 20%. In these soils, calcium forms the predominant exchangeable base, constituting 60 to 80% of the total exchangeable cation.
The predominance of exchangeable calcium give rise to Ca- clay which imparts a neutral reaction to the soil. The pH value varies from 6.5 to 7.5. When the proportion of exchangeable hydrogen (H) is high it gives rise to acid soil. In such soils, exchangeable calcium is correspondingly low, and in highly acid soils it is almost absent. In such cases the clay is saturated with hydrogen cations (H+) and forms H-clay. Acid soils are less fertile. It is called base unsaturated soil.
When exchangeable sodium form more than 10 to 15% of the total exchangeable cation it gives rise to alkaline soils. The pH value of such soils is usually greater than 8.0. When the proportion of exchangeable sodium exceeds these limits (or saturates the colloidal complex), the clay is turned into a Na-clay.
The soil is now highly alkaline and the pH value ranges from 9 to 12 Alkaline soils are also Jess fertile. Soils with a high calcium base saturation are in the most satisfactory physical and nutritional condition. A calcium dominated soil is granular in structure and ensure good drainage and aeration.
(b) Type of colloid:
Type of colloid affects the cation exchange. Montmorillonite colloid hold the calcium ion with greater tenacity than Kaolinite at a given base saturation. As a result, Kaolinite will liberate calcium much more readily than Montmorillonite.
(c) Associated ions:
Presence of exchangeable calcium in excessive quantities in a soil will limit the availability of potassium to plants. In same manner, high-exchangeable potassium may depress the availability of potassium to plants. In same manner, high- exchangeable potassium may depress the availability of magnesium.
(d) Adsorption of cations:
Colloidal clay (humus) hold in varying amount of plant nutrients (calcium, magnesium, potassium, nitrogen, phosphorus and most of the micronutrients) which are available to plant.
(e) Property of base exchange:
Base exchange (cation exchange) property checks leaching losses of available nutrients. On application of potassium sulphate fertilizer in the soil, potassium ions are held on the surface of colloids by cation exchange process. Subsequently, exchangeable potassium ions are directly available to plants.
Cation Exchange and Soil Fertility:
Cation exchange capacity is the best-index of soil fertility. By cation exchange, hydrogen ions from the root hairs and microorganisms replace nutrient cations from the exchange complex. The nutrient cations are forced into the soil solution where they can be assimilated by the adsorptive surface of roots and soil organisms, or they may be removed by drainage water.
(i) Cation saturation and Soil fertility:
Soil with a high calcium base saturation are the most satisfactory physical and nutritional condition. A calcium-dominated soil is granular in structure and porous. Calcium-dominated clay ensures good aeration and good drainage, thus increases fertility of the soils.
Base unsaturated soils are acidic in nature due to exchangeable hydrogen. These soils are less fertile. Base saturated soils with dominant sodium cations are alkaline in nature. Alkaline soils are not fertile due to de-flocculation, stickiness, hard to work, poor drainage and poor aeration.
(ii) Cation exchange and Soil fertility:
Due to the property of cation exchange (base exchange) the soluble inorganic fertilizer nutrients are not washed away from the soil. For example, ammonium sulphate fertilizer is added to the soil, ammonium ions are held on the surface of colloids by cation exchange. Ammonium ions are taken up by plants. This process checks nutrient losses by leaching and make the soil fertile. The cations Ca, Mg, K, and NH4 are held on the colloidal surfaces and are readily available to plants.
(iii) Influence of complementary adsorbed cations and soil fertility:
The order of strength of adsorption, when the ions are present in equivalent quantities, is as follow:
Al3+> Ca2+> Mg2+> K+ = NH4+> Na+
Consequently, a nutrient cation such as K+ is less tightly held by the colloids if the complementary ions are Al3+and H+ (acid soils) than if they are Mg++ +Na+ (neutral to alkaline soils). The loosely held K+ ions are more readily available for absorption by plants or for leaching in acid soils.
There are also some nutrient “antagonisms”, which in certain soil cause inhibition of uptake of some cations by plants. Thus, potassium uptake by plants is limited by high levels of calcium in some soils. Likewise, high potassium levels are known to limit the uptake of magnesium even when significant quantities of magnesium are present in the soil.
Anion Exchange:
The process of anion exchange is similar to that of cation exchange. Under certain conditions hydrous oxides of iron and aluminium show evidence of having positive charges on their crystal surfaces. The positive charge of colloids are due to addition of hydrogen (H+) in hydroxyl group (OH–) resulted in net positive charge (OH2+). This + charge will attract anions (—).
The capacity for holding anions increases with the increase in acidity. The lower the pH the greater is the adsorption. All anions are not adsorbed equally readily. Some anions such as H2 PO4– are adsorbed very readily (quickly) at all pH values in the acid as well as alkaline range. Cl– and SO4– ions are adsorbed slightly at low pH but none at neutral soil, while NO3– ions are not adsorbed at all. Hence, at the pH commonly prevailing in cultivated soils—nitrate (NO3), chloride (Cl) and sulphate (SO4) ions are easily lost by leaching.
In general, the relative order of anion exchange is:
OH > H2PO4–>SO4–>NO3–
Importance of Anion Exchange:
The phenomenon of anion exchange assumes importance in relation to phosphate ions and their fixation. The exchange is brought about mainly by the replacement of OH ions of the clay mineral.
The reaction is very similar to cation exchange:
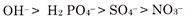
{kind=link}
The adsorption of phosphate ions by clay particles from soil solution reduces its availability to plants. This is known as phosphate-fixation. As the reaction is reversible, the phosphate ions again become available when they are replaced by OH ions released by substances like lime applied to soil to correct soil acidity.
Hence, the fixation is temporary. The whole of the phosphate adsorbed by clay is, however, not exchangeable, as even at pH, 7.0 and above. So, substantial quantities of phosphate ions are still retained by clay particles.
The OH ions originate not only from silicate clay minerals but also from hydrous oxides of iron and aluminium present in the soil. The phosphate ions, therefore, react with the hydrous oxides also and get fixed as in the case of silicate clay, forming insoluble hydroxy-phosphates of iron and aluminium.

{kind=link}
If this reaction takes place under conditions of slight acidity it is reversible, and soluble phosphate is again liberated when hydroxy-phosphate comes in contact with ions. If the reaction takes place at a low pH under strongly acid conditions, the phosphate (ions) are irreversibly fixed and the totally unavailable for the use of plants.
===================================
===================================
Chemistry and Behaviour of Phosphorus Present in Soil
Chemistry of Phosphorus:
1. Sorption Reactions:
The surfaces on which phosphate ions enter into sorption reactions of two types-surfaces of constant charge e.g. crystalline clay minerals and surfaces of variable charge including Fe3+ and Al—oxides and organic matter where H+ and OH– ions determine the surface charge and calcite (CaCO3) in which Ca2+ and CO ions involve the charge development.
Besides, some other clay minerals including amorphous such as allophane also involves in the phosphate sorption.
Hydrated Fe and Al oxides are the most important surfaces of variable charge in most soils excepting peats and highly calcareous soils. These oxides have surfaces of negatively charged OH groups which take up and dissociate protons (H+) and hence they are amphoteric having either negative, zero or positive charge depending on pH.
The pH at which there are equal numbers of positive and negative charges on the surface is known as point of zero charge (PZC). At pH levels below the PZC, phosphorus and other anions like SO42- and H3SiO4– are attracted to the positively charged oxide surfaces.
2. Precipitation Reactions:
Precipitation reactions mainly govern by the solubility product principles which are controlled by the pH of the system.
When some common phosphatic fertilizers like super phosphate, mono ammonium phosphate, Di-ammonium phosphate, some poly phosphates etc. are applied to the soil, within a very short time the released soluble phosphorus converts into very less soluble forms rendering unavailable and with time passes the strong insoluble phosphate fertilizer reaction products will form depending on the nature and type of soil as well as soil reaction.
In acid soils mono-calcium phosphate produces a number of substances like di-calcium phosphate (dihydrate and anhydrate), CaFe2 (HPO4)4. 8H2O; CaAl H(PO4)2.6H2O etc. whereas in calcareous soils, di-calcium phosphate (CaHPO4) is the dominant initial reaction product and in presence of excess amounts of calcium carbonate (CaCO3), octacalcium phosphate may also form.
Further, when di-ammonium phosphate is applied to soils, the following reaction products viz. Ca4 (PO4)3.3H2O; Ca2 (NH4)2 (NPO4)2.2H2O, CaHPO4-2H2O; CaNH4PO4.H2O; CaxH2 (PO4)6-5H2O etc. will form. Dicalcium phosphate dihydrate is one of the most dominant reaction products formed in high-calcium soils followed by octacalcium phosphate.
When polyphosphate fertilizers are applied to soils it undergoes precipitation and adsorption reactions. In addition the orthophosphate present initially plus which formed by the hydrolysis of polyphosphates react with the soil components similar to that happened in orthophosphate compounds.
Hydrolysis of polyphosphates results in a stepwise breakdown forming orthophosphates and different short chain polyphosphate fragments. Then such short chain polyphosphates undergo further hydrolysis. However, reactions of polyphosphates in soil and the nature of substances produced are dependent upon the rate of their reversion back to orthophosphates.
Slow rate of hydrolysis permits condensed phosphates to sequester or form soluble complexes with soil cations and hence reduce phosphate retention in soils. Two mechanisms namely chemical and biological are involved in the hydrolysis of polyphosphates. In soils, where both mechanisms can function, the rate of hydrolysis will be rapid.
{kind=link}
Enzymatic activity is the most important factor which controls the rate of hydrolysis. Phosphatases associated with plant roots and rhizosphere organisms are believed to be responsible for biological hydrolysis of pyro-and polyphosphates. Various factors like, temperature, soil pH, moisture, organic carbon content etc. can affect the transformation of polyphosphates.
Behaviour of Phosphorus:
Both organic and inorganic forms of phosphorus undergo transformation in soils leading to either release or retention of phosphorus. It is evident that decomposition of organic phosphorus substances gives both active and inactive substances.
The active substances are primarily the portions of the residues that have not yet been transformed into microbial products, whereas the inactive forms of phosphorus behave similarly to the resistant forms of nitrogen in humic acid.
1. Organic Phosphorus:
During mineralisation of organic phosphorus substances, the release of inorganic phosphorus takes place in the soil solution and such released phosphorus reacts very quickly with various soil components forming insoluble complex phosphatic compounds and there by unavailable to the plants.
Mineralisation of organic phosphorus is of three types:
(i) Based on the lowering of organic phosphorus level in soils due to long term cultivation.
(ii) Based on the results of short laboratory investigations decreasing the level of organic phosphorus with simultaneous increase in the amount of inorganic phosphorus in the soil and
(iii) Based on monitoring levels of soil organic phosphorus in the presence and absence of plants considering seasonal variation.
Mineralisation of organic phosphorus is carried by phosphatase enzymes and these enzymes are broad group of enzymes which catalyze the hydrolysis of both esters and anhydrides of phosphoric acid. However, there are a wide range of micro-organisms that are capable of mineralising (dephosphorylating) organic phosphorus on soils through their phosphatases activities.
Phosphatase activity of a soil is due to the combined functioning of the soil micro-organisms and any free enzymes present. Mineralisation of organic phosphorus is not entirely similar to that of organic carbon and nitrogen mineralisation and the mineralisation of organic phosphorus increases with an increase in soil pH but organic carbon and nitrogen mineralisation did not.
Most of the organic soil phosphates are present as inositol phosphate esters and these are prone to adsorption resulting less available in soils having higher adsorption capacity. The ultimate process by which organic phosphates are rendered available is by cleavage of inorganic phosphate by means of a phosphatase reaction.
The principle of this reaction is hydrolysis which is shown below:

{kind=link}
For carrying out the mineralisation of organic phosphatic substances in soils it is essential to have some idea about C: N: P ratios in the soil. A carbon: nitrogen: phosphorus (C: N: P) ratio of 100: 10: 1 for soil organic matter has been advocated, but its values ranges from 229: 10: 0.39 to 71: 10: 3.05—depending on nature and type of soils.
C: P inorganic ratio – Process Operates
200: 1 or less – Mineralisation
Above 200: 1 but – Neither net mineralisation nor
Less than 300: 1 – Net immobilisation
300: 1 and above Immobilisation
A concentration of about 0.2% phosphorus is critical in the mineralisation of organic phosphorus substances. If the system contains less than this, net immobilisation takes place, as both the plant and the native soil phosphorus are utilised by micro-organisms. The transformation of P takes place both in upland (aerobic) and low land submerged (anaerobic) soils.
2. Inorganic Phosphorus:
It is evident that most of the soluble inorganic phosphorus either released from the mineralisation of organic phosphorus or applied as soluble phosphatic fertilizers are rendered unavailable to the plants and hardly 20% of the applied phosphatic fertilizers are available to the plant.
The reasons for such recovery are the conversions of soluble form of phosphorus to a form which is very less soluble through reactions with various soil components involving different mechanisms.
Such mechanism for the removal of phosphorus from the solution phase in the soil is known as “retention or fixation”. However, the retention of phosphorus in the soil involves various mechanisms namely, sorption and precipitation reactions.
=========================================================
=========================================================
3 Main Forms of Potassium in Soils
Form # 1. Soil Solution Potassium:
It is recognised as the readily available form of potassium to the plants. The potassium availability in soils is controlled not only by the soil solution potassium but also by its buffering capacity (ability of a soil to maintain potassium intensity). The soil solution potassium (intensity, I) is maintained by the exchangeable potassium (quantity, Q) in a dynamic equilibrium.
The higher dQ/dl or the higher potassium buffer capacity indicates that during active period of crop growth, the potassium concentration in the soil solution will be depleted very rapidly. Soil solution potassium content usually higher in arid region and saline soil ranging from 3 to 156 ppm whereas the content of the same is lower in humid region soils ranging from 1 to 80 ppm.
Concentration of water soluble potassium may be as low as 8 ppm in deficient soils. However, under actual field conditions, the potassium concentration of soil solution varies with concentration and dilution processes brought about by evaporation and rainfall respectively.
The potentiality of soil solution K for plant growth and nutrition is influenced by the presence of other cations like Ca, Mg, and Al in acid soils and Na in salt affected soils. The activity ratio of potassium at equilibrium (AReK) with respect to these above cations is a measure of the “intensity” of labile potassium in the soil indicating instantly available to plant roots.
Soils having same AReK values may have different capacity in maintaining AReK during depletion of K by crop uptake or leaching and hence for the K status of soils it is necessary to specify not only the status of potassium in the labile pool but also the way in which the intensity depends on the amount (quantity) of labile potassium present.
However, the detail discussion about the Q/I relationship of K is presented in the following section.
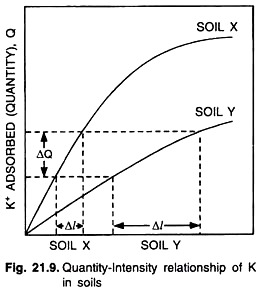
Buffer capacity indicates how intensity varies with quantity. A simple relationship between K intensity and K quantity for two soils (Soil X and Soil Y) having differential K adsorbing capacity is being depicted in Fig. 21.9. From the figure it is found that for both soils increasing intensity is accompanied by an increase in quantity.
{kind=link}
Soil X, however, shows a steeper rise in the slope than that of soil Y. Where an equal amount of K is removed from both soils by plants a similar decrease in the quantity of (∆Q) takes place. The consequent decrease in intensity (∆l), however, varies greatly for both soils (∆Ix and ∆IY).
This example shows that the two soils differ in their capacity of replenishing the soil solution with K. Soil X is better able to maintain the K concentration in the soil solution. Soil X is, therefore, more buffered than soil Y.
In quantitative terms the buffer capacity is expressed as the ratio ∆Q/∆I as follows:
BK = ∆Q/∆I,
where BK = buffer capacity of K in soils
The higher the ratio of ∆Q/∆I, the more the soil is buffered. Usually, the rate of K uptake by plant roots is higher than the diffusive flux of K towards the roots. The K concentration at the root surface may decrease during the period of plant uptake.
Such decrease in K concentration is dependent on the K buffering capacity of the soil. If the buffer capacity is high, the decrease may be low because of efficient K replenishment of the soil solution.
Again, for spoils having poor K buffer capacity, the concentration of K at the root surface may decrease appreciably throughout the plant growth period. For optimum growth of the plant, the concentration of nutrients in soil solution should be maintained above a certain level.
This concentration is termed as the critical nutrient concentration (CNC) below which the yield of crop is decreased. The critical level of K in the bulk soil solution is related to the buffer capacity of K (buffer power). The critical concentration is higher, the lower K buffer capacity.
In addition to AReK in assessing soil solution K, electro-ultra filtration (EUF) technique, a process of combination of electro dialysis and ultrafiltration, is used most satisfactorily for the characterisation of soil solution K (intensity) particularly in upland soils. The principle of EUF technique consists of utilizing the acceleration imposed upon ions by an electrical field for the separation of ions from soil colloids.
By adequate variation of voltage (50, 200 or 400 V) and timing (0-35 minutes), the total extractable K or any other nutrients can be separated into their water soluble and exchangeable forms with varying bonding energies.
Extraction and fractionation is done automatically. For the EUF-K fraction I (potassium in the extract obtained after 10 min of EUF—the first 5 min at 50 V and the next 5 min at 200V) and total EUF-K fractions (sum of all potassium fractions obtained after 35 min of EUF— the first 5 min at 50 V, the next 25 min at 200 V, and the last 5 min at 400 V).
The EUF-K fraction I considered as soil solution K (intensity factor) whiles the total EUF-K fractions as total amount of effectively available K (quantity factor). This electro- ultra filtration technique is better suited than that of AReK in distinguishing soils of varying K availabilities.
The amount of K in the soil solution is very low to meet the demand of the crop throughout the growing period and therefore it is necessary for satisfactory potassium nutrition of crops the soil solution K must be continuously replenished from the exchangeable, non-exchangeable and mineral forms of K.
Form # 2. Exchangeable Potassium:
Potassium ion (K+) is held by soil colloids through electrostatic attraction similar to other cations. However, potassium held by soil colloids is easily displaced or exchanged when extracting the soil with neutral salt solutions. The amount of K exchanged varies with cations and usually neutral normal ammonium acetate solution is used for the purpose.
A small amount of potassium in this fraction occurs in soils (<1.0% of the total potassium). The distribution of potassium on soil colloids as well as soil solution depends upon nature and amounts of complementary cations, anion concentration and nature and characteristics of clay minerals.
As for an example, if a soil colloid is saturated with potassium and in that condition a neutral salt like calcium sulphate is applied then the following exchange reaction takes place:

{kind=link}
Besides, if a soil is saturated with Al and Ca and in that conditions the application of muriate of potash gives the following exchange reaction:

{kind=link}
When muriate of potash is applied to soils containing adsorbed calcium and aluminium, calcium is more easily replaced than aluminium by potassium. Coarse textured sandy soils having a greater base saturation lose very little of their exchangeable potassium by leaching as compared to soils containing low basic cations.
Liming is considered as the most common method of increasing the base saturation of soils which results the decrease in the loss of exchangeable potassium.
Sites for K Exchange:
It is evident that the exchangeable potassium on soil colloids is not homogeneous. Usually potassium is held at three binding sites of soil colloids namely p-(planar) position (outer surface of colloids, non-specific), e-(edge) position and i-(inner or inter layer) position (specific for K).
The amount of K held on p-position is in equilibrium with the soil solution K, while the amount of soil solution K in equilibrium with K held on e and i positions of soil colloids is low. However, under actual field situations, potassium concentrations in the soil pollution are probably the net result of three possible equilibria.
It is evident that the exchangeable form of potassium plays an important role in replenishing soil solution potassium removed by either intensive cropping or leaching losses.
In view of the above fact, it is very much essential to establish the quantity relationship between exchangeable K (Q quantity) and the activity of potassium in the soil solution (I intensity) in order to assess the availability of more labile potassium in soils to plants (Fig. 21.10).

{kind=link}
The Q/I concept has been developed by Beckett which is used for predicting the status of potassium in soils. Different parameters of the above curve have some practical implications in relation to potassium in soils and plants.
∆K = Amount through which the soil gains or loses potassium in bringing equilibrium (Q, quantity factor).
ARK = Activity ratio of potassium (I, intensity factor).
AReK = Activity ratio of potassium at equilibrium
∆Kex = Exchangeable or labile pool of potassium
KSP = Specific sites for potassium
PBCK = Potential buffering capacity
ARK (Intensity Factor, I):
It is calculated from the determined concentration of calcium, magnesium, potassium and sodium correcting to the appropriate activities with the help of extended Debye-Huckel theory.
AReK (Activity Ratio of K at Equilibrium):
It is a measure of availability or intensity of labile pool of potassium in soil and can be modified by potassium fertilization, being increased due to application of K fertilizers. However, the availability of potassium in soils can either be increased or decreased due to liming which modifies the AReK values either favorably or adversely.
∆Kex:
It is used more successfully for the estimation of labile soil potassium held in plannar (p) positions. Greater values of labile potassium i.e. more negative (-Kex) indicate a higher potassium release into the soil solution which results greater amount of potassium in the labile pool. However
the application of potassic fertilizers and lime in the cropped field have been found to be increased the amount of potassium in the labile pool.
Ksp (Specific Sites for Potassium):
It is a curved portion of the Q/I relationship while the linear portion of the curve (Q/I) is attributed to non-specific sites for potassium. Specific sites having high affinity for potassium are believed to exist on edges of clay minerals (e-positions) and in interlayer or wedge zones of weathered micas (i-positions).
Whereas non-specific sites for potassium are associated with planar surfaces of clay minerals (p-positions). The i-position has the greatest specificity for K+ which largely account for K+ fixation in soils.
PBCK (Potential Buffering Capacity of Potassium):
It is a measure of ability of a soil to maintain potassium concentration in the soil solution. The potential buffering capacity for potassium is proportional to the cation exchange capacity (CEC) of the soil that means, with an increase in CEC the value of PBCK increases and vice-versa resulting from changes in ARK values.
A high PBCK value indicates a good potassium supplying power of soils whereas a low PBCK value signifies very low potassium supplying power of soils indicating frequent potassium fertilization. However, the higher PBCK value may be obtained due to time application which probably as a result of increase in pH-dependent cation-exchange capacity.
If PBCK is low, small changes in exchangeable potassium produce large differences of potassium content in the soil solution. This value is very small coarse textured sandy soils where mainly organic matter is contributed to the CEC value. In such soils, extensive leaching, rapid plant growth etc. deplete available potassium within a few days.
In general, the relation between exchangeable and soil solution potassium is a good measure of the availability of the labile pool of potassium in soils to plants. The ability of a soil to maintain the activity ratio of K against depletion by crop uptake, leaching etc. is controlled by nature of the labile pool potassium as well as the rate of release of fixed potassium and diffusion and transport of K+ ions in the soil solution.
Form # 3. Non-Exchangeable and Mineral Form of Potassium:
Potassium in these forms is not readily available to the plants. However, non-exchangeable potassium pools not instantly available to plants, can contribute significantly to the maintenance of the labile pool of potassium in the soil. On the other hand, in some soils these fractions of potassium may become available as water-soluble and exchangeable forms are removed by leaching, crop uptake etc.
These forms of potassium are consisting of different K-bearing minerals namely primary minerals (K-feldspars) and micas (muscovites, biotites etc.), originating from the parent rock and secondary minerals (clays of the illitic group) formed by alteration of micas.
The main source of K+ for plants growing under natural conditions is from the weathering of K containing minerals mentioned above. In potash feldspars, potassium occurs in the interstices of the Si, Al—O framework of the crystal lattice and held rigidly by covalent bonds. The weathering of feldspars starts at the surface of the particle.
Initially potassium is released by water and weak acids at a more rapid rate, However, with the progress of weathering, a Si—Al—O residue envelope is formed surrounding the un-weathered core. This layer reduces the rate of potassium loss from the mineral and hence protects K from further degradation.
Minerals of the mica type and also the secondary minerals of 2: 1 layer silicates vary in structure from feldspars and thereby these minerals also differ in their properties of releasing and binding potassium.
The micas consist of unit layers each containing two Si, Al—O tetrahedral sheets between which is an M (Al, Fe, Mg)—O, OH octahedral sheet potassium (K+) ions occupy the approximately hexagonal spaces between the unit layers and as a result the distance between unit layers is relatively small i.e. 1.0 nm in micas.
The replacement of un-hydrated interlayer K+ by hydrated cations like Na+, Ca2+ or Mg2+ expands the mineral with an increase in the distance between the unit layers i.e., 1.4 nm in vermiculite (Fig. 21.11 and 21.12).

{kind=link}

{kind=link}
Usually K+ of the lattice is vulnerable to weathering and can diffuse out of the mineral in exchange for other cations. High H+ concentrations and low K+ concentrations in the soil favour the net release of non-exchangeable, inter layer K+.
This K+ release may be an exchange process associated with diffusion in which K+ adsorbed to i-positions of the inter layer zone is replaced by other large cations like Na+, Ca2+ and Mg2+ resulting an expansion of clay lattice and the formation of wedge zones (See in above figure).
“Frayed edge” or “wedge” zone formation is typical of weathering micas which results release of interlayer K+. The rate of release of K+ by weathering not only depends on the K content of the mineral, but also affected by structural variation between minerals.
The gradual release of potassium from positions of mica lattice results in the formation of illite (hydrous mica) and eventually vermiculite with accompanying gain of water or H3O+ and swelling of the lattice (Fig. 21.13).
{kind=link}
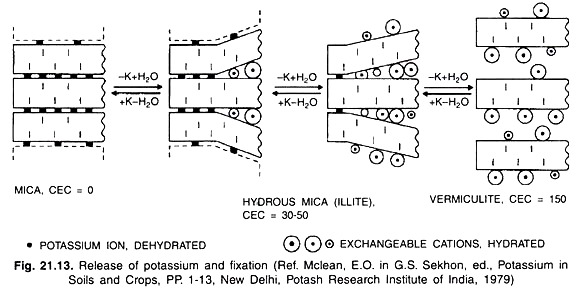
There is also an increase in specific surface charge and CEC of clay minerals formed during the weathering of K containing minerals as well as transformation of mica. However, the applied soluble potassic fertilizers are converted to fixed or non- exchangeable forms of K and such conversions are affected by various factors. Besides fixation, the applied potassic fertilizers also undergo leaching loss from soils.
It is evident that a substantial amount of potassium can be lost through leaching in soils containing more amounts of sands due to flooding. However, in case of silty loam and clay loam soils, the loss of K through leaching is less because of fairly higher rate of adsorption of potassium by soil colloids. Again, in organic soils e.g. muck soils have high exchange capacities.
The bonding strength for cations like potassium is not great and the amount of exchangeable K tends to vary with the intensity of rainfall. Therefore, care should be taken for the supply of potassium to crops through its annual application.
Leaching losses can be reduced with the application of lime to the soil by maintaining a favourable pH level. Leaching losses of potassium frequently occurs in coarse textured sandy or organic soils particularly in areas of high rainfall.